– Zandelisib Demonstrated 70.3% Objective Response Rate and 35.2% Complete Response Rate –
– 87.5% of Responses Achieved in First Two Cycles of Therapy, 75% of Complete Responses in first four cycles –
– 9.9% of Patients Discontinued Therapy Due to a Drug Related Adverse Event –
– Primarily, Grade 3 AESIs to Date Occurred in Cycles 1-3. –
MEI Pharma, Inc. (NASDAQ: MEIP), a late-stage pharmaceutical company focused on advancing new therapies for cancer, and Kyowa Kirin Co., Ltd. (Kyowa Kirin, TSE: 4151), a global specialty pharmaceutical company creating innovative medical solutions utilizing the latest biotechnology, today announced that data from the zandelisib clinical development program, including the Phase 2 TIDAL study evaluating the intermittent dosing of zandelisib, an orally administered investigational phosphatidylinositol 3-kinase delta ("PI3Kδ") inhibitor in clinical development for the treatment of B-cell malignancies, is highlighted in a poster and an oral discussion session at the European Hematology Association 2022 Hybrid Congress.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20220610005042/en/
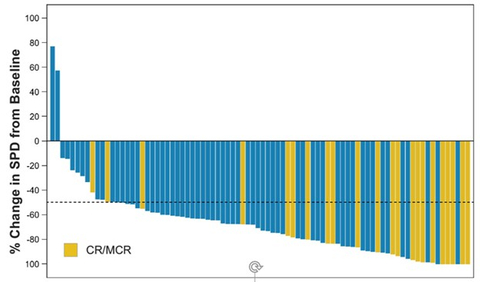
Best Change in SPD from Baseline** 7 patients not included because they did not have post-baseline imaging scans. (CR, complete response; MCR, metabolic complete response; SPD, sum of the product of the longest perpendicular diameters). (Graphic: Business Wire)
“While many patients with indolent non-Hodgkin’s lymphomas, such as follicular lymphoma, achieve durable responses with current standard of care therapies, patients generally continue to need multiple treatments throughout the course of their disease due to relapse, necessitating the development of novel treatment options,” stated Professor Wojciech Jurczak, M.D., Ph.D. Department of Clinical Oncology, Maria Sklodowska-Curie National Research Institute of Oncology in Kraków, Poland, presenter of the Phase 2 TIDAL study oral presentation and the Phase 3 COASTAL study principal investigator. “I believe the data presented at EHA this year are very exciting and supportive of the zandelisib development program, particularly as we continue to enroll the Phase 3 COASTAL study evaluating zandelisib in combination with rituximab.”
Clinical Data from the Phase 2 TIDAL Study Evaluating Zandelisib in Patients with Relapsed or Refractory Follicular Lymphoma
Oral Presentation Title: Efficacy and Safety of Zandelisib Administered by Intermittent Dosing (ID) in Patients with Relapsed or Refractory (r/r) Follicular Lymphoma: Primary Analysis of the Global Phase 2 Study TIDAL
Session Title: Indolent & mantle cell non-Hodgkin lymphoma – Clinical
Presenter: Wojciech Jurczak, PhD, MD
Time: June 11, 11:30am-12:45pm CEST
Abstract Code: S208
Study Details
The ongoing TIDAL study (NCT03768505) is a global, open-label Phase 2 trial evaluating zandelisib as a single agent in two disease cohorts: one in relapsed or refractory (r/r) follicular lymphoma (FL) and one for r/r marginal zone lymphoma (MZL), in both cases after failure of at least two prior systemic therapies, including chemotherapy and an anti-CD20 antibody. Patients were administered zandelisib once daily for two 28-day cycles as response induction regimen, followed thereafter by once daily dosing for the first seven days of each subsequent 28-day cycle, an intermittent dosing (ID) regimen. The ID regimen was introduced to potentially allow Treg repopulation and to mitigate immune-mediated adverse events of special interest (AESI) without loss of disease control.
Enrollment in the FL cohort is complete; enrollment in the MZL cohort is ongoing. The FL cohort enrolled a total of 121 patients, with the first 91 consisting of the primary efficacy population (PEP) for the evaluation of overall response rate (ORR) and duration of response (DOR).
The median age of patients enrolled with FL was 64 years old. Enrolled patients were generally heavily pretreated; the median number of prior therapies was 3 (range 2-8) and 96% of patients had received prior chemoimmunotherapy. Further, 28 patients (23%) received prior stem cell transplant, 50 patients (41%) were refractory to last therapy, 41 patients (34%) had tumors ≥5 cm, and 68 patients (56%) were POD24 (progression of disease within 24 months of prior chemoimmunotherapy).
The primary efficacy endpoint is ORR as assessed by an independent review committee (IRC) using a modified Lugano criteria. The data cutoff date is September 30, 2021, approximately 6 months after the last patient in the primary efficacy population (PEP) received their first dose of zandelisib. Data from the enrolling MZL cohort is not reported.
Efficacy
The primary endpoint of ORR of zandelisib as a single agent in the PEP was 70.3% (N=64) as assessed by IRC; the complete response rate was 35.2% (N=32). Responses across sub-groups at high risk were all >63%:
- Refractory to last treatment: 64.3%
- >2 prior therapies: 63.3%
- POD24 (progression of disease within 24 months): 66.7%
The ORR represents the primary endpoint of the TIDAL study.
Responses were first observed in the first two cycles of therapy in 87.5% of all response (N=56), and 75% of all CRs (N=24) were observed in the first four cycles. The disease control rate (CR+PR+SD) was 85%. As of the data cutoff date, the data are not sufficiently mature to accurately estimate the final DOR in the PEP. An additional data cut is planned for approximately 14 months after the last patient in the PEP is enrolled to evaluate DOR and provide more mature safety results.
Safety and Tolerability
With a median follow-up of 9.4 months (range 0.8-24) in the safety population of 121 pts, 9.9% (N=12) of patients had discontinued zandelisib due to any drug-related adverse event. Adverse events occurring in ≥ 10% of 121 patients regardless of relationship to zandelisib were: diarrhea, neutropenia, nausea, fatigue, abdominal pain, pyrexia, decreased appetite, constipation, rash and anemia.
Grade 3 (AESI were diarrhea in 5% (N=6), colitis in 1.7% (N=2), cutaneous rash in 3.3% (N=4), stomatitis in 2.5% (N=3), and 0.8% (N=1) each for AST and ALT elevation, and non-infectious pneumonitis. Primarily, Grade 3 AESIs to Date Occurred in Cycles 1-3. No Grade 4 or Grade 5 AESI were recorded.
Treatment-emergent COVID-19 infections (any grade) were reported in 8.3% (N=10) of patients, and 8.3% (N=10) of patients reported Grade 3 infections. Four COVID infections were fatal, as were 1 case each of pneumonia and tumor lysis syndrome.
Updated Clinical Data from the Phase 1B Study Evaluating Zandelisib in Patients with Relapsed or Refractory Follicular Lymphoma
Poster Title: Zandelisib on Intermittent Dosing as a Single Agent or in Combination with Rituximab or Zanubrutinib in Relapsed or Refractory (r/r) Follicular Lymphoma (FL): Results from a Multi-Arm Phase 1b Study
Session Title: Indolent & mantle cell non-Hodgkin lymphoma – Clinical
Time: June 10, 2:30pm-3:45pm CEST
Presenter: Jacob Drobnyk Soumerai, MD
Abstract Code: P1114
Study Details
The ongoing Phase 1B trial (NCT02914938) is an open-label, multi-arm, study evaluating zandelisib alone, in combination with rituximab or with zanubrutinib in about 160 patients with R/R chronic lymphocytic leukemia (CLL) or small Lymphocytic Lymphoma (SLL) or B-cell lymphoma. The presentation reports on the 69 patients enrolled with relapsed or refractory (r/r) follicular lymphoma (FL) across three groups:
- Group 1: Zandelisib single agent 60 mg once daily for two 28-day cycles then on the intermittent dosing (ID) schedule of 60 mg once daily on days 1-7 in cycles ≥3
- Group 2: Zandelisib (same schedule as Group 1) in combination with rituximab 375 mg/m2 weekly x 4 in cycle 1 and on Day 1 of cycles 3-6
- Group 3: Zandelisib 60 mg on ID from cycle 1 and zanubrutinib 80 mg twice daily
Zandelisib administration was continued until disease progression or intolerance. Patients received a median of 2 prior therapies and 65% were POD24.
Safety and Tolerability
With a median duration of drug exposure of 17.1 months in Group 1, 20.5 months in Group 2, and 7.1 months in Group 3, six patients (8.7%) discontinued therapy due to treatment related adverse events. Grade 3 AESI, ranging from 0% to 11% across all groups, were: diarrhea/colitis, AST/ALT increase, rash, infection and atrial fibrillation (with zanubrutinib only). No grade 4-5 adverse events of special interest were reported. One patient in Group 3 had reversible grade 4 drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. Grade 3-4 neutropenia was observed in 11 pts (16%).
Excluding COVID-19, 7 patients (10.1%) had a grade 3 infection (2 had appendicitis, and 1 each had infectious diarrhea, CMV colitis, pneumonia, soft tissue infection, and tooth infection). COVID-19 was reported in 4 patients, fatal in 2: one with zandelisib alone and 1 with zandelisib-rituximab. No other Grade 5 events were reported.
Efficacy
The overall response rate in the 65 evaluable patients was 84.6% (55/65) and the complete response rate was 24.6% (16/65). The overall response rate was 77.8% (14/18) in Group 1, 94.7% (18/19) in Group 2, and 82.1% (23/28) in Group 3.
The median duration of response in the heterogeneous patient groups with small patient populations was: 31.1 months in Group 1, 25.8 months in Group 2, and not yet mature in Group 3. The median drug exposure of 17.1, 20.5, and 7.1 months, respectively.
“The data reported at the 2022 European Hematology Association demonstrate the potential of zandelisib, an investigation clinical candidate employing a unique intermittent dosing schedule, to provide a high response rate in a well-tolerated manner either as a single-agent or in combination with other therapies,” said Richard Ghalie, M.D., chief medical officer of MEI Pharma. “We remain encouraged by zandelisib’s potential as we continue its development using the intermittent schedule as an oral, chemotherapy-free, time-limited, therapeutic option to meet the needs of patients.”
“The data presented at the EHA congress support zandelisib’s potential value as a single agent or combination therapy,” said Yoshifumi Torii, Ph.D., Executive Officer, Vice President, Head of R&D Division of Kyowa Kirin. “We continue to work with MEI Pharma to conduct global clinical trials with the goal of providing innovative treatment options for patients with B-cell malignancies.”
About Zandelisib
Zandelisib, a selective PI3Kδ inhibitor, is an investigational cancer treatment being developed as an oral, once-daily, potential therapeutic option for patients with B-cell malignancies. Clinical trials are investigating the efficacy and safety of zandelisib as a single agent and in combination with other modalities while administered on an Intermittent Dosing Regimen (ID) and in a time-limited manner when dosed in combination. The ID leverages molecular and biologic properties specific to zandelisib.
In November 2021, MEI Pharma and Kyowa Kirin announced data from ongoing Phase 2 TIDAL study (NCT03768505) evaluating zandelisib as a single agent for follicular lymphoma (FL) patients who received at least two prior systemic therapies. Zandelisib demonstrated a 70.3% objective response rate (ORR) as determined by an Independent Review Committee (IRC) assessment in the primary efficacy population (n=91). In addition, 35.2% of patients achieved a complete response. At the time of the data cutoff, the data were insufficiently mature to accurately estimate duration of response (DOR). In line with previously reported data from the Phase 1B study, zandelisib was generally well tolerated. With 9.4 months (range: 0.8-24) median duration of follow-up in the total study population (n=121), interim data demonstrated a discontinuation rate due to any drug related adverse event of 9.9%. Patients enrolled in the study will continue to be followed for safety and DOR.
Ongoing zandelisib studies include the cohort in TIDAL evaluating patients with r/r marginal zone lymphoma (MZL) and continuing evaluation of the cohort of patients in the study with r/r FL. Also ongoing is the Phase 3 COASTAL study (NCT04745832), comparing zandelisib plus rituximab to standard of care chemotherapy plus rituximab in patients with r/r FL or MZL who received more than one prior line of therapy, which must have included an anti-CD20 antibody in combination with chemotherapy or lenalidomide. COASTAL, which is also evaluating time-limited intermittent administration of zandelisib, is intended to support marketing applications in the U.S. and globally.
Other ongoing studies include a Phase 2 pivotal study in Japan (NCT04533581) in patients with indolent B-cell non-Hodgkin's lymphoma (iB-NHL) without small lymphocytic lymphoma (SLL), lymphoplasmacytic lymphoma (LPL), and Waldenström's macroglobulinemia (WM) conducted by Kyowa Kirin.
In March 2020, the FDA granted zandelisib Fast Track designation for the treatment of adult patients with r/r follicular lymphoma who have received at least two prior systemic therapies. In November 2021, the FDA granted zandelisib Orphan Drug designation for the treatment of patients with follicular lymphoma.
In April 2020, MEI and Kyowa Kirin entered a global license, development, and commercialization agreement to further develop and commercialize zandelisib. MEI and Kyowa Kirin will co-develop and co-promote zandelisib in the U.S., with MEI booking all revenue from the U.S. sales. Kyowa Kirin has exclusive commercialization rights outside of the U.S.
About Follicular Lymphoma
Follicular lymphoma (FL) is the most common indolent lymphoma, comprising about 20-30% of all non-Hodgkin lymphomas (NHL). The disease also forms on B cells, is chronic in most cases and tends to progress slowly. Most people diagnosed with FL are over 65 years of age. Sometimes follicular lymphomas can transform into a more aggressive form of large B-cell lymphoma, a fast-growing (aggressive) type of NHL.
About MEI Pharma
MEI Pharma, Inc. (Nasdaq: MEIP) is a late-stage pharmaceutical company focused on developing potential new therapies for cancer. MEI Pharma's portfolio of drug candidates contains multiple clinical-stage assets, including zandelisib, currently in ongoing clinical trials which may support marketing approvals with the U.S. Food and Drug Administration and other regulatory authorities globally. Each of MEI Pharma's pipeline candidates leverages a different mechanism of action with the objective of developing therapeutic options that are: (1) differentiated, (2) address unmet medical needs and (3) deliver improved benefit to patients either as standalone treatments or in combination with other therapeutic options. For more information, please visit www.meipharma.com. Follow us on Twitter @MEI_Pharma and on LinkedIn.
About Kyowa Kirin
Kyowa Kirin strives to create and deliver novel medicines with life-changing value. As a Japan-based Global Specialty Pharmaceutical Company with a heritage of 70+ years, we apply cutting-edge science including an expertise in antibody research and engineering, to address the needs of patients and society across multiple therapeutic areas such as Nephrology, Oncology, Immunology/Allergy and Neurology. Across our four regions – Japan, Asia Pacific, North America and EMEA/International – we focus on our purpose, to make people smile, and are united by our shared values of commitment to life, teamwork/Wa, innovation, and integrity. You can learn more about the business of Kyowa Kirin at: https://www.kyowakirin.com.
Forward-Looking Statements
Under U.S. law, a new drug cannot be marketed until it has been investigated in clinical studies and approved by the FDA as being safe and effective for the intended use. Statements included in this press release that are not historical in nature are "forward-looking statements" within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995, including statements regarding the results of our clinical trials of zandelisib, the anticipated timing of our submission of an FDA marketing application for zandelisib, the anticipated timing of the disclosure of the final study data for our Phase 2 TIDAL trial, the timing and success of enrollment for our Phase 3 COASTAL trial, our projected financial position and our expected cash runway, the overall advancement of our product candidates in clinical trials and our plans to continue development of our product candidates. We may in some cases use terms such as “predicts,” “believes,” “potential,” “continue,” “anticipates,” “estimates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “likely,” “will,” “should” or other words that convey uncertainty of the future events or outcomes to identify these forward-looking statements. You should be aware that our actual results could differ materially from those contained in the forward-looking statements, which are based on management's current expectations and are subject to a number of risks and uncertainties, including, but not limited to, our failure to successfully commercialize our product candidates; the availability or appropriateness of utilizing the FDA’s accelerated approval pathway for our product candidates; final data from our pre-clinical studies and completed clinical trials may differ materially from reported interim data from ongoing studies and trials; costs and delays in the development and/or FDA approval of our product candidates, or the failure to obtain such approval, of our product candidates; uncertainties or differences in interpretation in clinical trial results; the risk that our clinical trials are discontinued or delayed for any reason, including for safety, tolerability, enrollment, manufacturing or economic reasons; the impact of the COVID-19 pandemic on our industry and individual companies, including on our counterparties, the supply chain, the execution of our clinical development programs, our access to financing and the allocation of government resources; our inability to maintain or enter into, and the risks resulting from our dependence upon, collaboration or contractual arrangements necessary for the development, manufacture, commercialization, marketing, sales and distribution of any products; competitive factors; our inability to protect our patents or proprietary rights and obtain necessary rights to third party patents and intellectual property to operate our business; our inability to operate our business without infringing the patents and proprietary rights of others; general economic conditions; the failure of any products to gain market acceptance; our inability to obtain any additional required financing; technological changes; government regulation; changes in industry practice; and one-time events. We do not intend to update any of these factors or to publicly announce the results of any revisions to these forward-looking statements.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220610005042/en/
Contacts
MEI Pharma Contacts:
David A. Walsey
MEI Pharma
Tel: 858-369-7104
investor@meipharma.com
Jason I. Spark
Canale Communications for MEI
Tel: 619-849-6005
jason.spark@canalecomm.com
Kyowa Kirin Contacts:
Hiroki Nakamura (Global)
Corporate Communications Department
media@kyowakirin.com
Stacey Minton
SVP, Corporate Affairs (EU)
comms@kyowakirin.com